巴西醫療器械注冊與計算機軟硬件及外圍設備制造流程融合解析
在當今全球化的醫療健康領域,巴西作為拉丁美洲的重要市場,其醫療器械監管體系嚴謹而復雜。隨著醫療科技的飛速發展,計算機軟硬件及外圍設備在醫療器械制造中的應用日益廣泛,使得產品注冊流程呈現出跨領域融合的特點。本文旨在梳理巴西醫療器械注冊的核心流程,并探討計算機軟硬件及外圍設備制造在此過程中的關鍵角色與合規要點。
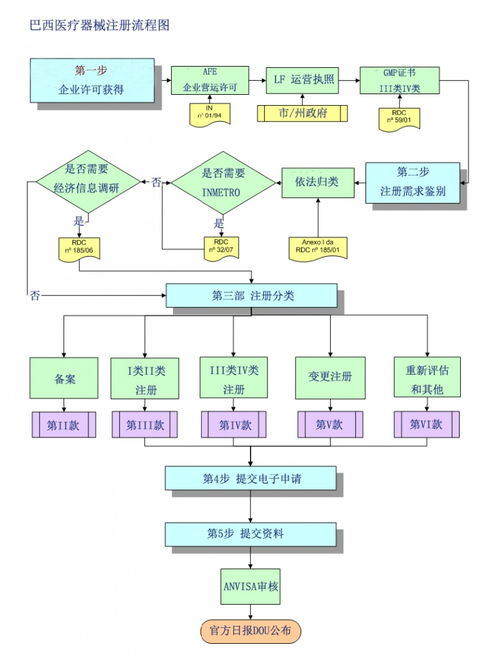
一、 巴西醫療器械注冊核心流程圖解
巴西的醫療器械監管主要由國家衛生監督局(ANVISA)負責,其注冊流程可概括為以下關鍵階段:
- 產品分類與路徑確定:
- 根據風險等級(I、II、III、IV類),將醫療器械劃分為不同類別,并確定相應的注冊路徑(如簡化注冊、常規注冊或特殊途徑)。
- 計算機軟硬件融合點:對于集成軟件(如SaMD-醫療器械軟件)或依賴特定硬件運行的設備,其風險分類需綜合考慮軟件功能、數據安全及硬件穩定性。
- 技術文件準備與質量管理體系:
- 編制詳盡的技術文件,包括產品描述、性能評估、臨床評價(如適用)、生物相容性測試、電氣安全與電磁兼容性報告等。
- 制造環節融合:制造商必須建立并維護符合ANVISA RDC 16/2013等法規要求的質量管理體系(QMS)。對于涉及計算機硬件制造(如醫療成像設備主機)或專用外圍設備(如傳感器、控制器)的生產,其設計控制、采購、生產、檢驗過程均需嚴格記錄并確保可追溯性。軟件作為醫療器械或其組成部分,其開發生命周期需遵循IEC 62304等標準,并納入QMS管理。
- ANVISA注冊申請提交與評審:
- 通過巴西指定的電子系統(如Pet)提交注冊申請及全套文件。
- ANVISA進行技術評審,可能要求補充信息或澄清。對于復雜產品,評審時間可能較長。
- 軟硬件合規性:評審將重點關注軟件驗證與確認報告、網絡安全(如數據加密、訪問控制)、硬件與軟件的接口兼容性、以及外圍設備的可靠性與安全性證據。
- 注冊批準與上市后監督:
- 獲得注冊號("Registro")后,產品方可合法在巴西市場銷售。
- 上市后需履行警戒義務,包括不良事件報告、定期安全性更新,并保持質量管理體系有效運行以應對ANVISA檢查。
- 持續制造合規:任何涉及軟硬件的設計變更或制造過程變更,均需評估其對注冊狀態的影響,必要時提交變更申請。
二、 計算機軟硬件及外圍設備制造的特殊考量
在支持醫療器械注冊的制造過程中,計算機制造領域需特別注意:
- 硬件制造與標準符合性:醫療設備中的計算機硬件(如嵌入式系統、服務器、工作站)常需符合額外的安全、可靠性和環境耐受標準(如IEC 60601-1系列標準)。制造過程應確保組件來源可靠、生產線校準精確,并進行嚴格的環境應力篩選和老化測試。
- 軟件開發與生命周期管理:作為醫療器械核心的軟件,其需求分析、架構設計、編碼、測試、部署和維護的全過程必須文檔化并驗證。對于人工智能/機器學習驅動的軟件,還需考慮算法透明度和持續學習模型的管理策略。
- 外圍設備集成與接口安全:外圍設備(如輸入輸出設備、存儲單元、網絡模塊)的制造需確保其與主設備的物理和邏輯接口穩定、安全。數據傳輸的完整性與保密性至關重要,特別是涉及患者健康信息時需符合巴西數據保護法(LGPD)的要求。
- 供應鏈與網絡安全:全球化供應鏈背景下,硬件組件(如芯片、傳感器)和軟件庫的來源安全性需嚴格審核。制造環境及產品本身應具備抵御網絡攻擊的能力,防止因惡意篡改導致的安全風險。
三、 流程整合與成功關鍵
成功在巴西注冊一款融合先進計算技術的醫療器械,關鍵在于早期規劃與跨領域協作:
- “法規前置”策略:在產品設計與制造規劃初期,即邀請法規專家介入,確保軟硬件設計從一開始就瞄準ANVISA的合規要求。
- 質量管理體系一體化:將軟件工程(如敏捷/DevOps)與硬件制造的質量管理流程無縫整合到統一的QMS中,確保端到端的質量控制與可追溯性。
- 本地化支持:考慮聘請巴西本地合規代表("Responsável Técnico")或咨詢機構,他們熟悉ANVISA的評審習慣和溝通方式,能有效橋接技術語言與法規語言。
- 持續合規與創新平衡:在快速迭代的技術環境中,制造商需建立靈活的變更管理機制,既能及時響應市場與技術更新,又能確保每一步變更都處于法規框架之內。
巴西醫療器械注冊并非單一的行政程序,而是一個將產品安全有效性、質量管理與特定國家法規深度結合的系統工程。對于高度依賴計算機軟硬件及外圍設備的現代醫療器械,制造商必須將卓越的工程技術、嚴謹的制造管控與前瞻性的法規策略融為一體,方能順利叩開巴西市場的大門,并為全球患者提供安全可靠的創新醫療解決方案。
如若轉載,請注明出處:http://www.nnqprdh.cn/product/79.html
更新時間:2026-05-28 10:31:59